Cardiovascular disease (CVD) remains a leading cause of death and morbidity worldwide. According to the WHO, 18 million of the 68 million deaths annually can be attributed to it. That's more than 1 in 4. This does not include those whose quality of life is severely impacted by it. Despite the positive impact of LDLc-lowering drugs (statins, PCSK9 inhibitors), blood-lowering, anti-diabetic, and weight-loss medications, other risk factors remain to be addressed.
Among them is lipoprotein little a [Lp(a)], an independent and causal risk factor for cardiovascular disease. Except for a few with extremely high Lp(a) who were lucky enough to be diagnosed, often after family members encountered cardiovascular events early in life, it has been largely met with a shrug in general medicine. The dynamic, however, is quickly changing now as the PCSK9 outcomes trials are strongly supportive that lowering Lp(a) should be able to lower CVD risk (O'Donoghue et al 2019) and new strategies mostly centered around sequence-targeted therapeutics allow for robust Lp(a) reductions.
This week, Novartis disclosed that the eagerly anticipated results from the pivotal trial of the most advanced of these clinical candidates, the RNaseH antisense compound pelacarsen has been delayed. It was originally projected to deliver results in 2025, but is now guided to read out in the first half of 2026. As this has raised a few eyebrows, I will take the opportunity to provide an overview of the Lp(a) competitive landscape and the pharmaceutical companies involved.
Lp(a) is a genetic determinant of CVD risk independent of LDLc and ApoB100
Lp(a) is quite similar to LDL-cholesterol lipoprotein particle, but in addition to carrying one molecule of apolipoprotein B 100, cholesterol and phospholipids in its outer shell, there is a apolipoprotein (a) molecule attached to ApoB100 via a disulfide bond. If LDL cholesterol is referred to as the ‘bad’ cholesterol, Lp(a) on a per particle basis is the lipoprotein supervillain. It not only gets more easily through the blood vessel endothelial cell layer so that it becomes a substrate for plaque build up, it is quite inflammatory as a result of being a good carrier of oxidized phospholipids. It is also thought that there may be a prothrombotic effect partly due to its similarity and evolutionary relationship with plasminogen.
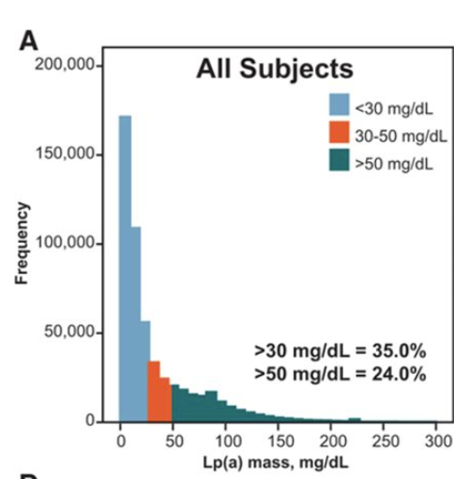
Unlike LDLc, Lp(a) levels are largely genetically determined and cannot be modified by nutrition and exercise. They stay more or less constant after 5 years of age. Levels range widely within a population. Whereas many have Lp(a) levels of 10mg/dL or less (~22nM), the distribution has a long tail with ~10% of the population, especially black people, who have more than 90mg/dL. This puts them at ~1.4x and higher risk of suffering CVD events like CVD death, myocardial infarction or of developing calcific aortic valve and peripheral artery disease (
Erqou et al 2009). The increased risk is maintained after having suffered a first event with almost 2x the risk for persons in the top decile versus those below 10mg/dL (
Madsen et al 2020). This is relevant when thinking about using Lp(a) lowering as a strategy for primary or secondary CVD prevention.
Evidence that intervention can reduce CVD risk
Currently the best advice one can give to somebody with highly elevated familial Lp(a) is to address all the other modifiable CVD risk factors. In terms of medication, this typically means LDLc-lowering by statins and, even better so, the newer PCSK9 inhibitors.
This is because PCSK9 inhibition reduces Lp(a) by 20-30% (Sabatine et al 2017; Schwartz et al 2018; Ray et al 2017) . The high variability of the Lp(a) reduction between subjects allowed for the determination that in the FOURIER trial of Amgen’s evolocumab there was a 15% relative risk reduction for certain cardiovascular events for each 25nM Lp(a) lowering (~11.6mg/dL; O'Donoghue et al 2019). Remarkably, those above the median Lp(a) at baseline (37nM) not only had the highest absolute Lp(a) reduction, they had a 23% lower CVD hazard ratio versus only 9% for those below the median. Importantly, these findings were independent of the magnitude of LDL-c lowering in the same subjects which was the same across the Lp(a) ranges.
This in my mind is the strongest evidence that not only life-long differences in Lp(a) have a meaningful impact on CVD risk, but even just 5 years of pharmaceutically lowering Lp(a) may have a big impact.
The Lp(a) competitive landscape
It is therefore not surprising that half a dozen or so notable Lp(a)-lowering efforts are in clinical development. At the same time, medical societies like the EAS, EHA and AHa and KOLs are busy raising awareness around Lp(a). Still, a lot of work remains to be done: less than 1% in the western medical systems have been tested, despite the fact that guidelines are increasingly recommending getting tested at least once in a lifetime. While universal numbers are hard to come by, reports from various health systems suggest that testing is finally gaining traction (e.g. Bhatia et al 2023). The pump is thus getting primed for the successful roll-out of Lp(a)-lowering agents.
RNaseH pelacarsen (Ionis, Novartis)
Leading the pack is pelacarsen, originally discovered and developed by Ionis Pharmaceuticals and subsequently licensed to Novartis in 2019. Pelacarsen is a subcutaneously administered GalNAc-ASO working by RNaseH-mediated knockdown of apo(a) mRNA. It is currently in a global pivotal phase 3, Lp(a)HORIZON outcomes study evaluating its ability to reduce major cardiovascular events (MACE) in a secondary prevention setting in 8000+ patients with Lp(a) >70mg/dL.
The 80mg once monthly should achieve 80% Lp(a) lowering based on a phase IIb study in a similar population (Tsimikas et al 2020). Safety and tolerability have been good. This means an absolute reduction of ~55-60mg/dL for a patient at the lower end of the range, a number that has been estimated (Madsen et al 2020) to be enough for ~20% relative risk reduction.
Given the uncertainties about the exact relationship between pharmaceutical Lp(a) lowering and risk reduction, Novartis conservatively added a second cut analyzing the effect of pelacarsen in the population with >90mg/dL of baseline Lp(a) for efficacy. If the study hits in either cohort, it can be considered successful.
The study is an event-driven one, meaning that in this case 993 MACE need to accumulate. The fact that the data monitoring committee (DMC) has apparently communicated to Novartis that the projected results are now due in 2026 instead of 2025 suggests that insufficient MACE events have occurred since study start in 2019 and completion of enrolment in mid-2022 (2 ½ years minimal follow-up).
It is plausible that the slower-than-anticipated event rate has to do with a population that is highly controlled for other risk factors, especially LDLc, as I expect most to be on maximally tolerated statins and a good proportion on PCSK9 therapeutics, too. More optimistically, slow event accrual could also be due to an extraordinary efficacy of pelacarsen. I suspect, however, that the trial protocol provided for interim looks not only for futility (early looks), but also for efficacy at this late stage of the trial. The most likely scenario therefore is that the DMC saw maybe 800 events and concluded that the study would benefit from running its full course. At the very least, the study was not stopped for futility.
Personally, 70mg/dL is too low a baseline Lp(a) to expect a strong MACE response. I see Lp(a)-lowering to have most initial utility in the top Lp(a) decile (>90-100mg/dL) and it would therfore not surprise me if HORIZON only hit in that bracket.
RNAi olpasiran (Arrowhead, Amgen)
Hot on the heels of pelacarsen is olpasiran, an RNAi agent discovered by Arrowhead Pharmaceuticals, licensed to and now in development by Amgen. This liver-targeted RNAi trigger commenced a pivotal phase 3 trial in another secondary prevention study for atherosclerotic cardiovascular disease in 2022 and has finished enrolling 7000+ subjects in just 1 ½ years. This means that results from OCEAN(a) Outcomes could become available within a year of Lp(a)HORIZON in late 2026.
The apparent enthusiasm by Amgen is encouraging since they, as the developer of PCSK9 antibody Repatha, are best positioned to understand the likely impact of modulating Lp(a) on CVD outcomes. In addition, the efficacy of olpasiran trounces that of pelacarsen with a time-averaged Lp(a) reduction of more than 95% (vs -80%) despite of using quarterly instead of monthly subcutaneous dosing (O'Donoghue et al 2022). The marked difference in efficacy will also allow for insights into whether, as is widely assumed now and as is the case for LDLc, it is the absolute Lp(a) lowering that matters or the relative percent lowering.
Finally, I like the study because it focuses on a population in the top decile of baseline Lp(a) (>200nMol, ca >90mg/dL) where genetically CVD risk increases exponentially. I also would not be surprised if lowering Lp(a) well below the genetically defined 50mg/dL risk threshold brings further benefit in an interventional setting. Doing everything to keep these streaky macrophages from bursting by depriving them of their oxidized fatty meals could be quite beneficial.
RNAi lepodisiran (Dicerna, Eli Lilly)
Two years behind olpasiran in terms of phase 3 initiation, but every bit as potent and even more long-lived in its Lp(a)-lowering activity is RNAi rival lepodisiran. Lepodisiran is a Dicer-substrate RNAi molecule discovered by Dicerna (now Novo Nordisk) and developed by Eli Lilly.
 |
| Lp(a)-lowering by single dose of lepodisiran |
In a single dose study, a large dose of 608mg, achieved time-averaged ~95% reduction in Lp(a) over a year. A lower dose of 304mg effectively lowered Lp(a) by a peak ~98% in the first months before leveling off at around 90% at the half year mark (Nissen et al 2023).
The ACCLAIM-Lp(a) ASCVD phase 3 study in 12500 subjects in secondary prevention and for those at high risk will test 3 semi-annual doses before moving on to annual dosing. Though the dose has not been disclosed, I consider both 608mg and 304mg dosages possible with this dosing regime. The choice will have probably been strongly informed by whether Eli Lilly believes the clinical benefit of going from 10nM to 5nM Lp(a) will balance out formulation, drug administration, and potentially safety issues coming with the high dose.
Besides Eli Lilly, Novo Nordisk, Amgen, and pretty much everybody with a stake in the lipid-related CVD market believe in the advantages of infrequent drug administration as evidenced by their investments in this area. Novartis this quarter saw the PCSK9-targeting RNAi drug Leqvio (licensed from Alnylam) cross the $1B run-rate for the first time. It is 284mg of canonical siRNA oligonucleotide administered basically semi-annually. This demonstrates the shift from daily oral pills that most people will have stopped using after a year despite their need for life-long treatment. This is understandable since most of them do not suffer acute symptoms.
 |
| 2-dose study of zerlasiran |
This brings us to the final notable RNAi contender,
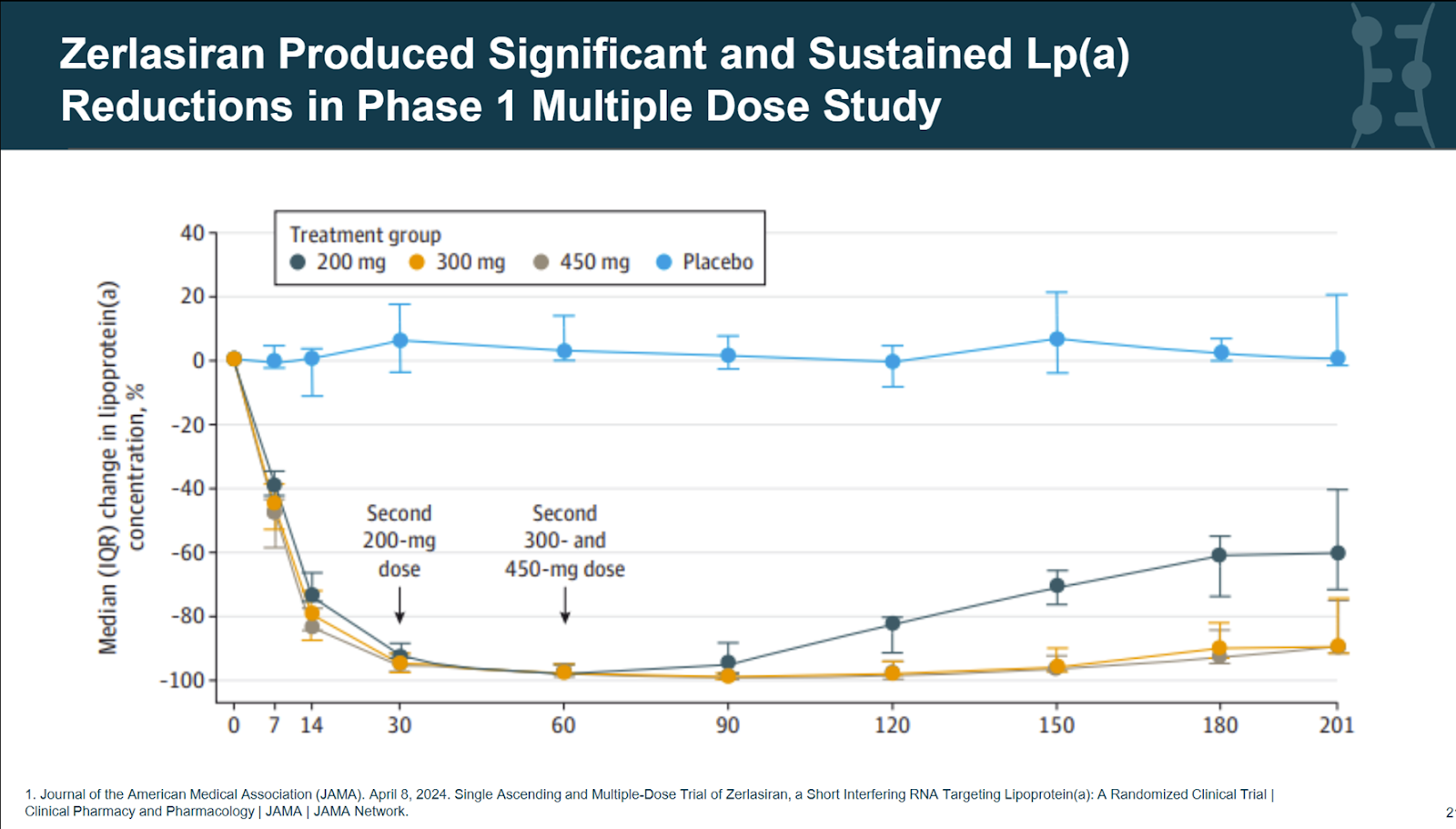
zerlasiran by Silence Therapeutics. In early trials, 300mg showed clear potential for maintaining Lp(a) reduction of more than 95% using quarterly dosing. Being phase 3 ready, Silence Therapeutics stood to benefit the most from a timely Lp(a)HORIZON reveal by Novartis. Not only may have they been able to adjust their Lp(a) target levels for their outcomes trial, a successful readout may also have been the trigger for a Big Pharma like Pfizer or Merck with no stake in Lp(a) yet to acquire zerlasiran, if not acquire Silence Therapeutics (market cap $300M) altogether.
Life-long Lp(a)-lowering using CRISPR genome editing
If you like infrequent dosing, why stop at semi-annual or annual when there are life-long options going after the same apolipoprotein(a) target? This includes base editor VERVE-301 by cardiovascular specialty company Verve Therapeutics which is partnered with Eli Lilly, but still in preclinical development. Already in phase I is competitor CTX-320 by CRISPR Therapeutics using more traditional CRISPR Cas9 cleavage of apo(a). Both use standard intravenous LNP delivery to liver hepatocytes.
Finally, in addition to RNAi and CRISPR, Eli Lilly is also developing an oral small molecule drug. Muvalaplin is designed to inhibit the interaction between ApoB100 and Apo(a) and thus the biogenesis of the Lp(a) particle. Daily doses barely lower Lp(a) by 60% and as a small molecule there are concerns about hemodynamic effects given the similarities of apo(a) and plasminogen (Nicholls et al 2023).
Disclosure: I have been and am still positioning myself for HORIZON by investing across the Lp(a) landscape, including Silence Therapeutics, Verve Therapeutics and Ionis. Focused Lp(a) testing companies would also fall into my interest area, so please pitch me ideas if you have some in this area.